ええ?なにこのポスト?
最近、Google社がタンパク質のフォールディングを計算できるAlphafoldをリリースした。AlphaFoldというと、2018年CASP13コンテストに参加したAlphaFold 1.0と2020年CASP14コンペティションんみ参加したAlphafold 2.0という二つのバージョンがある。Alphafold 1.0とAlphafold 2.0は、構造がまったく違っているので、このポスト両方を見てみるかと思う。
リソース
- Cryo-EMの利点について
- DeepmingからAlphafoldの一覧
- Alphafold 1.0の論文 (Natureで発表された)
- Alphafold 2.0の論文(準備中)
- Alphafold 2.0の論文 (上と違う)
- Alphafold 2.0ソースコード
- ニューラルネットワークにおけるAttentionの説明
- Tensorflow概要
- MSAについて詳しい説明
モチベーション
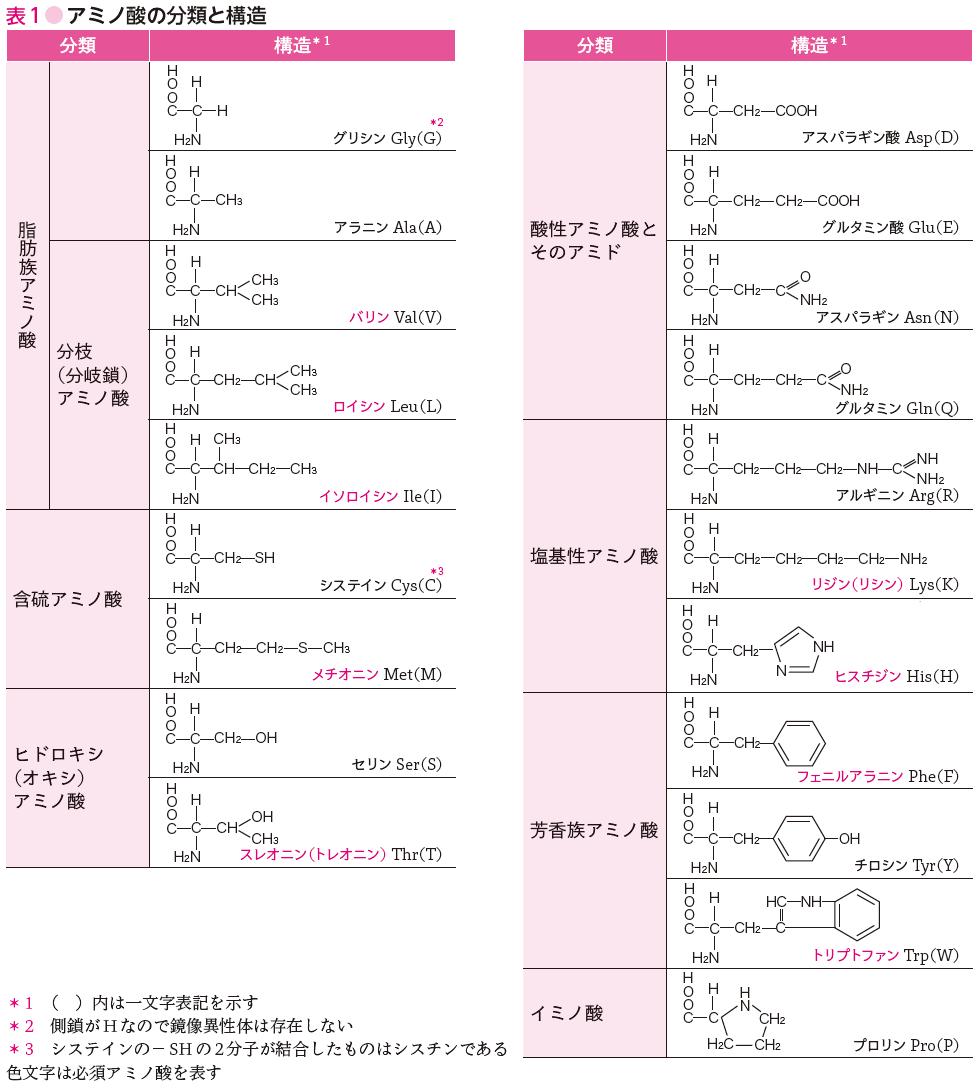
アミノ酸とは?
タンパク質は、アミノ酸のチェンです。また、ポリーぺプタイドとも呼ばれています。 人類の体の中、大体20種類のアミノ酸がある。
以下の点を注意しよう:
- アミノ酸をフールネーム、三つのローマ字、また一つのローマ字で表すことができます(例: グリシン = Gly = G)。
- チェンが始まった場所、NH2のある端末をN-terminusと呼ばれ、終わる場所、COOHのある端末をC-terminusと呼ばれます。つまり、方向性があります。。
- NH2とCOOHの間に一つの炭素があり、この炭素についている分子グループをSidechain,またRグループと呼ばれ、基本的にアミノ酸の性質を決める。
- Sidechain以外のグループをBackboneと呼ばれます。
- 真ん中の炭素原子をCαと呼ぶ
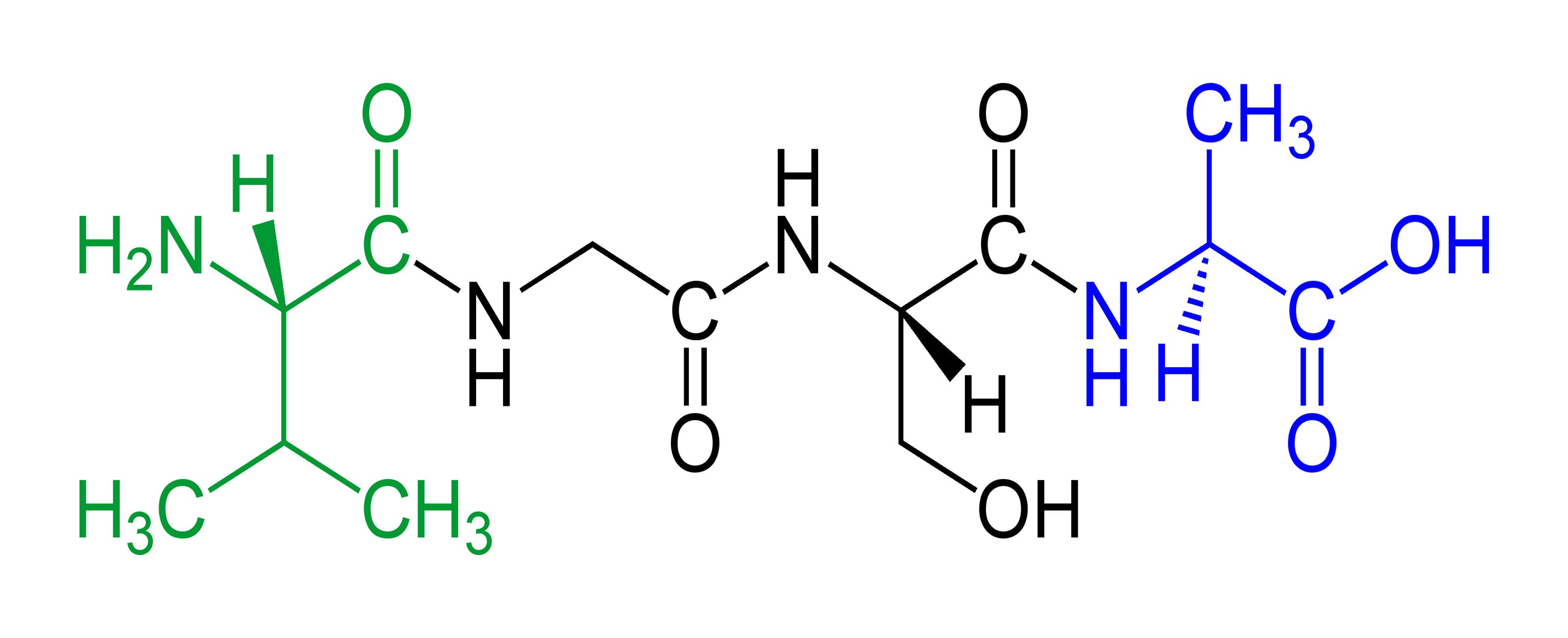
アミノ酸からタンパク質まで
アミノ酸で作られたタンパク質が特定の三次元の構造が持っていて、全般的な構造に影響を与えるのは、一次構造、二次構造、三次構造、四次構造である。
一次構造
一次構造は、アミノ酸の配列です。アミノ酸の配列は、遺伝子から直接に推定できるので、現在、人間のほとんどのタンパク質の配列既に解明しています。
二次構造
Rグループ以外のグループが水素結合により構造ができている。その中、αヘリックス、βシートが一番重要。水素結合とは、電子の分布が不均一によりできている静電的な結合で、アイオン結合・共有結合より圧倒的に弱い結合である。
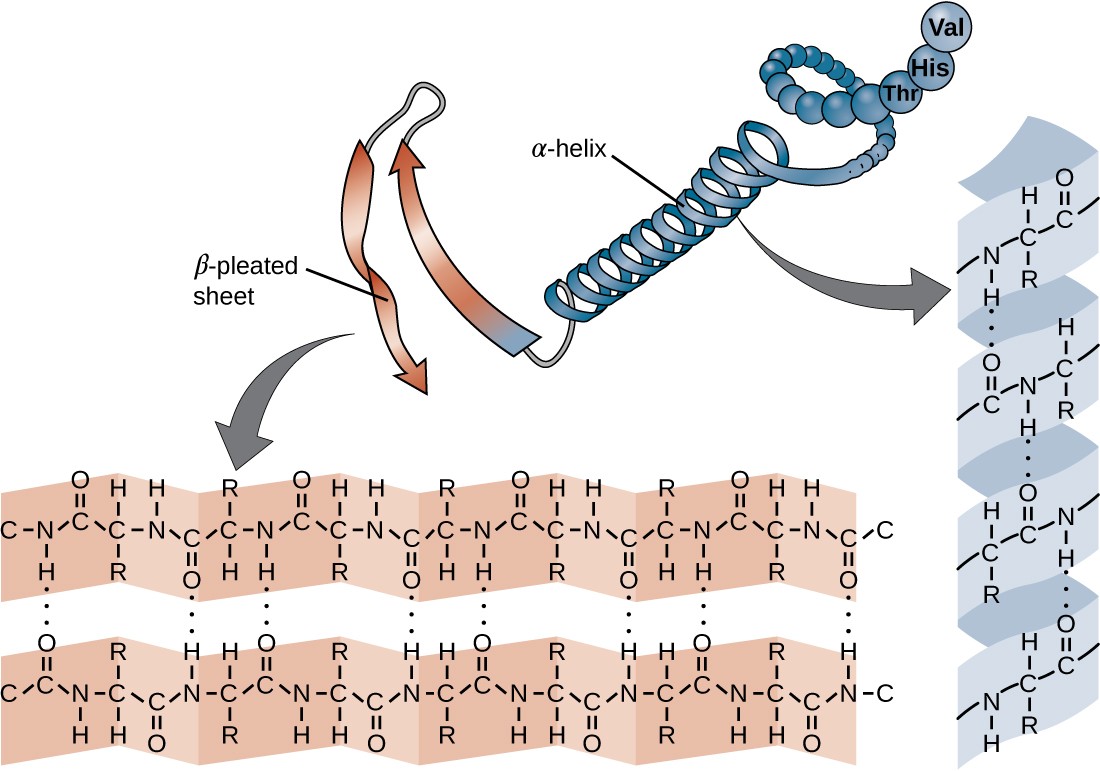
例として、実際の論文の中、このような図がよく見られる。
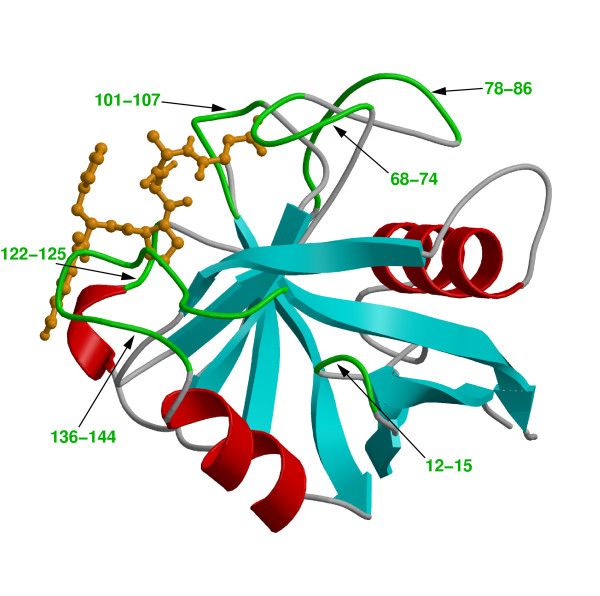
三次構造
三次構造は、Backbone, Sidechainの間の相互作用でできているものである。最も有名なのは二つのシステインのRグループ間にS-Sの共有結合(Sは硫黄元素、Disulphide-Bridge。共有結合は水素結合、アイオン結合よりも強い。また、タンパク質のからの安定さ・性能を影響するので、アルツハイマー病、ハンチントン病のようは病気、薬のデザインなどに関わっている重要な問題になっている。
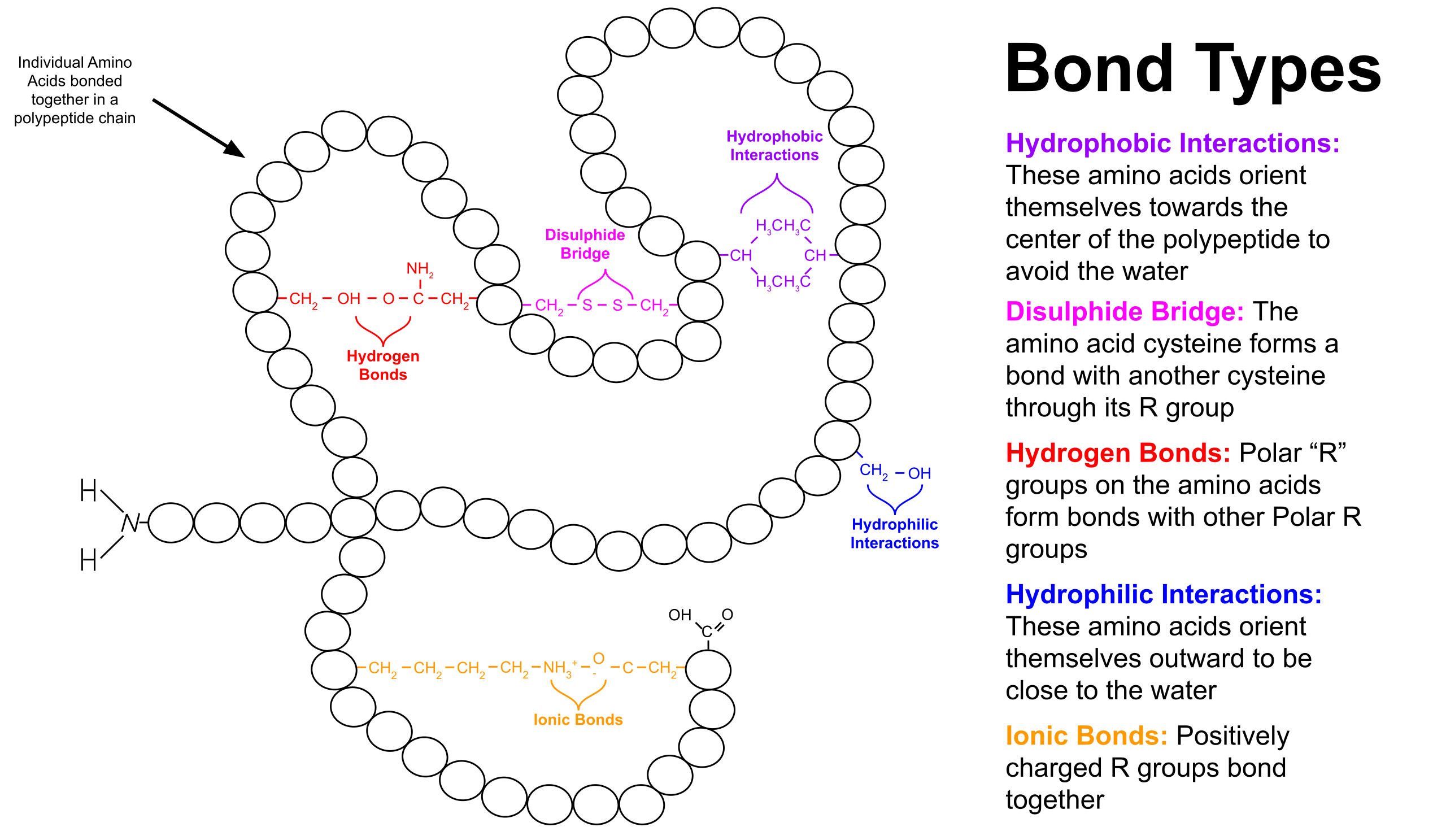
四次構造
四次構造は、複数の三次構造ができているアミノ酸のチェインの間の相互作用である。現在「一次構造がわかれば、三次構造は推定できる」という説が基本的に認めらている。しかし、「一次構造がわかれば、四次構造は推定できる」は争論中。
既存の構造解明手法
Alphafold 2.0で「タンパク質の構造を推定する」というと、一次構造からを三次構造することです。
Alphafoldは計算手法であり、今、計算結果を実験からできた結果と比べないといけない。現時点では、存在がわかっている2億のタンパク質の中、構造が解明されているのは~17万個。次、実験手法について紹介したいと思う。
X線結晶構造解析 (X-ray crystallography)
X線結晶構造解析は、結晶の原子および分子構造を決定する実験科学であり、結晶構造により入射するX線のビームが多くの特定の方向に回折する。これらの回折したビームの角度と強度を測定することにより、結晶学者は結晶内の電子密度の3次元画像を作成することができます。
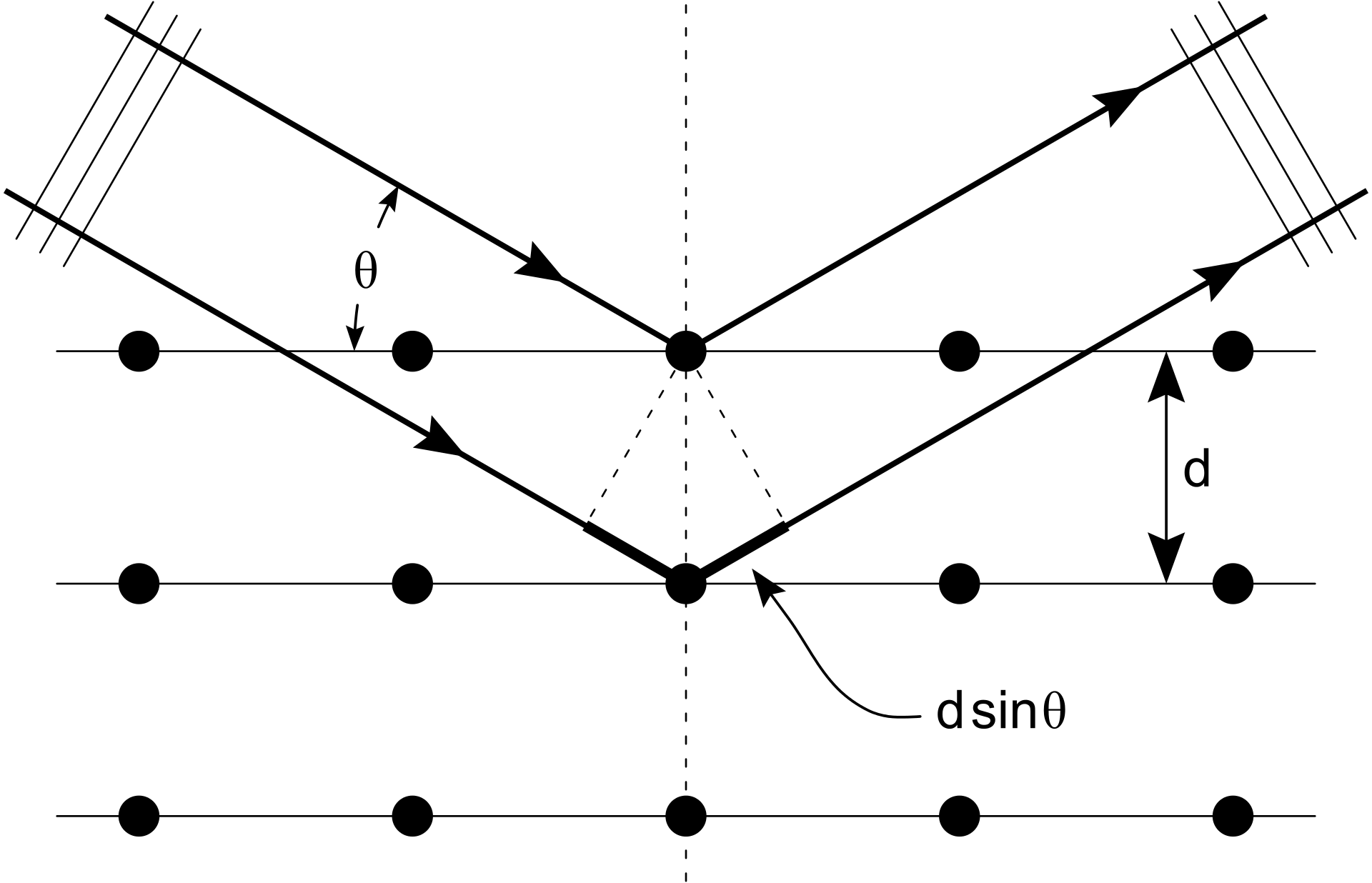
この手法は現時点でのGolden Standardと言える。しかし、タンパク質の結晶を手に入れるのは、実はものすごく難しいことです。その理由は若干があります:
- 水性溶液に過飽和までタンパク質を溶かす必要があります、しかし水に溶かしにくい膜タンパク質は、半分以上を薬のターゲットでありながら精製、結晶させるのは難しい。
- タンパク質のサンプル量があるほど必要です。
- 温度、pH、溶液の成分など、様々なファクターが最後の結果に影響を与えますので、工夫がかかってしまいます。
タンパク質の核磁気共鳴分光法 (Protein Nuclear Magnetic Resonance (NMR) Spectroscopy)
これはMRIの同じ原理、核磁気共鳴を用いる手法です。おおざっぱにすると以下の流れになります:
- 外部静磁場B0による、サンプルの原子核のスピンが一直線にする
- 一直線を振動磁界で(RF)で破壊する
- 原子核スピンの特性を分析する
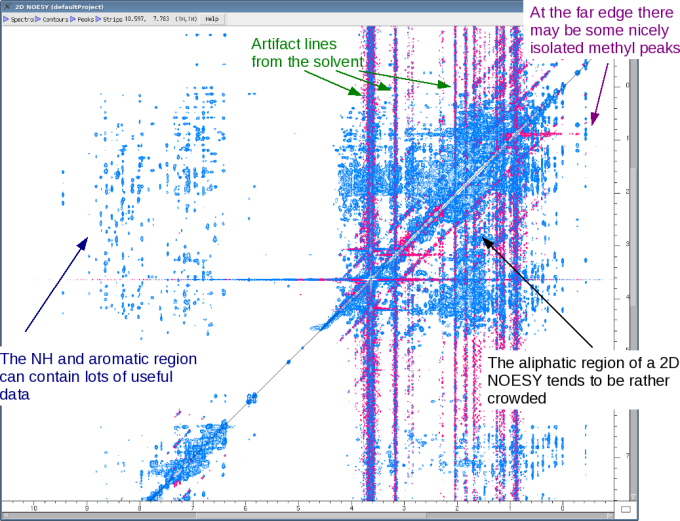
この手法にも欠点が若干あります:
- 装備は高いです(50万~5億円まで。解像度が良いほど)。
- 割と高いサンプル量が必要です。
- サンプルが大きな分子になると、ピークの数も増えてしまいますので、解明するのはも難しくなってしまう。
低温電子顕微鏡法 (Cryogenic electron microscopy, Cryo-EM)
Cryo-EMは透過型電子顕微鏡法の一種で、試料を低温(多くの場合液体窒素の温度)において解析する手法である。通常の電子顕微鏡での観察では、電子線による損傷と、高真空状態が生物試料に与える影響は大きかった。この手法では、氷の結晶が発生しないように一瞬でサンプルをフリーズして、電子でサンプルを調査できる
Cryo-EMは、以下の利点がある:
- 膜タンパク質構造でも解明できる
- タンパク質が「結晶できるか結晶できないか」
現時点では、これはAlphafoldの一番強い競争相手じゃないかと思う。データの傾向によると、2024年以降、Cryo-EMでできたタンパク質の構造の数がX線結晶に超えてゆく。
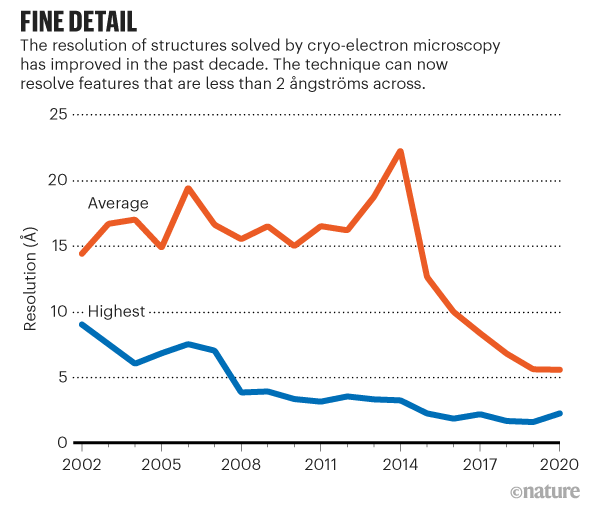
計算手法でタンパク質の構造を解明する共通
Critical Assessment of Techniques for Protein Structure Prediction (CASP) とは?
CASPとは、二年ごとにタンパク質を計算手法で解明するコンテストである。実験手法で解明されたばっかりタンパク質を回答として使って、タンパク質の構造を予測する計算手法を評価できる。
完全なタンパク質を予測するだけでなく、ある巨大なタンパク質の中、特定の機能を満たすドメイン(Domain)を予測することもある。
計算手法というと、主にシムレーション系とバイオインフォマティクス系という二つのアプローチがある。
シムレーションVSバイオインフォマティクス一覧
| 手法 | 概要 | 利点 | 不便 |
|---|---|---|---|
| シムレーション | アミノ酸のそれぞれの物理的な相互作用に注意する。熱力学・統計学・物理学に基づく手法 | 直接に構造を解くことができる | タンパク質のサイズが大きくなるほど予測は難しくなる |
| バイオインフォマティクス | タンパク質の遺伝子テンプレートを進化的な関係を調べる。バイオインフォマティクス・アルゴリズムに基づく手法 | 既に存在しているタンパク質データベース、遺伝子シーケンシングを効率的に利用できる。また、深層学習で相互関係を解析できる | 進化的な関係のある分子がない場合特に難しい |
Alphafoldがリリースされた前に、シムレーションでもバイオインフォマティクスでも満足できる予測を生成できなかった。
Alphafold1.0でも2.0でもバイオインフォマティクスのアプローチを利用している手法である。
CASPのカテゴリー
CASPは更にカテゴリーに分けている。毎回のカテゴリー少し調整されているけれど、基本的に以下のカテゴリーがある。
- Template-based Modeling (TBM): 既に解明されている更に似っているタンパク質がある。
- Free Modeling: 既に解明されている更に似っているタンパク質がない。
メトリックス
計算された構造を実験手法で手に入れた構造に比べると、以下のメトリックスを使う。
Angstrom Cα root-mean-square deviation at 95% residue coverage (Å r.m.s.d.95) 原子位置の平均二乗偏差
名前通り、このメトリックスは、計算された構造のアミノ酸のCαの位置を実験手法で得られた構造に比べる。単位はÅ (Angstrom), 10-10m(炭素の原子半径は0.7Å)。
また、95%とは、あるタンパク質の中、95%のアミノ酸のCαが揃えるために最低限の調整を意味している。
Local Distance Difference Test (LDDT)-Cα
原文によると、RMSDは複数の動きやすいドメインの評価において、大きいなドメインにバイアスが存在する。
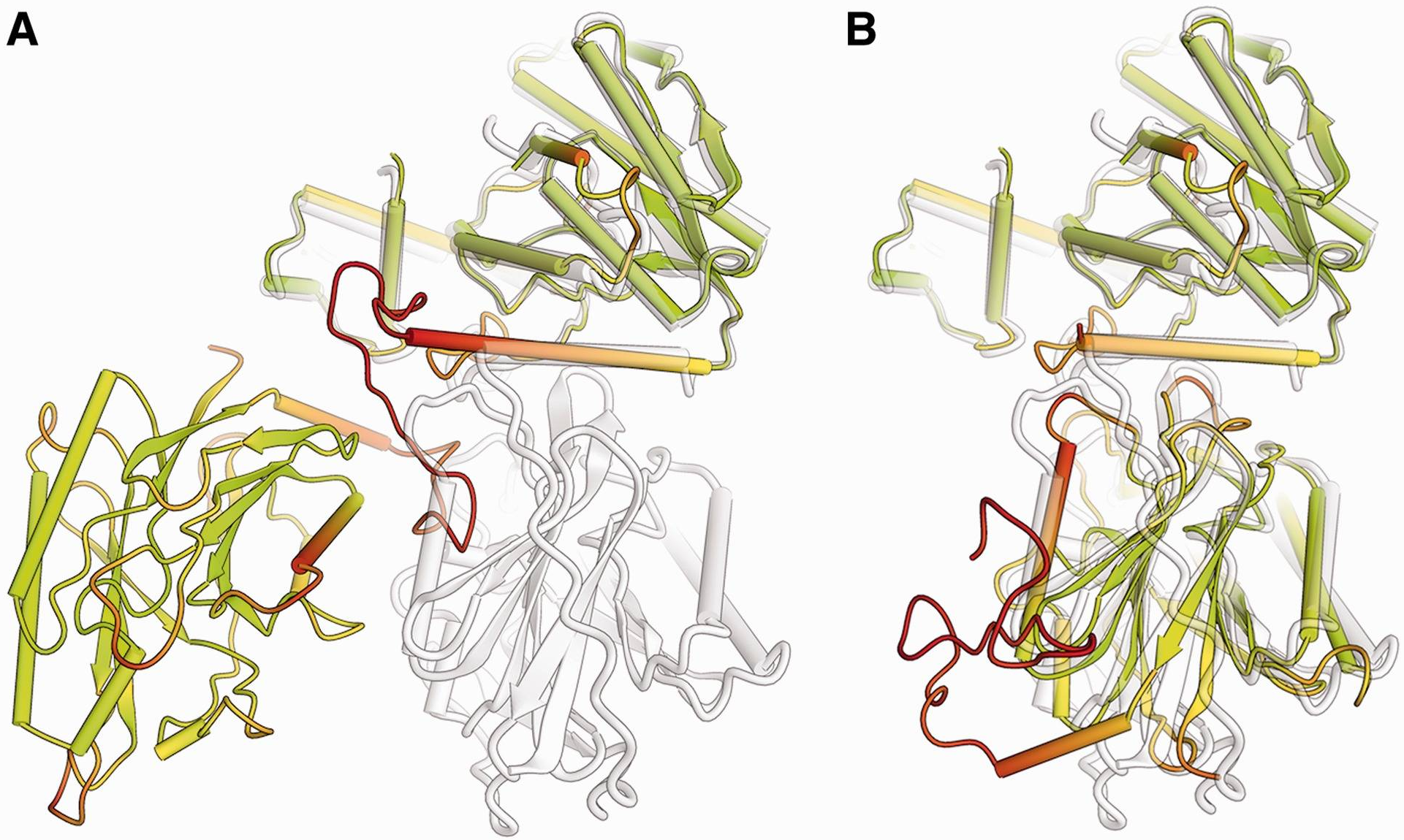
バイオインフォマティクス手法
Multiple Sequence Alignment (MSA) 多重整列
配列が似ているタンパク質・遺伝子・リボ核酸を揃えるアルゴリズムである。更に、MSAにおけるも深さ(Depth)という特徴量ある。これはニューラルネットワークの深さのことではなく、MSA行列における「一つの列における解明されているアミノ酸の数」の平均値である。

要素和
Alphafold 2.0 CASP14 (2020) 詳細
ハイレベル一覧
- 訓練ハードウエア:128 TPUv3コア、約2週間
- 訓練セット: 約17万公開されているタンパク質の構造
- テストセット:
- ロース:FAPE
- 結果:CASPドメインにおける0.96 Å r.m.s.d.95 (95%確信間隔 = 0.85 - 1.16Å)、二番目良いは2.8Å
- 構造:ステージ二つ:
- Evoformerニューラルネットワークブロック 48個
- MSA行列(s*r, s=シーケンスの数、r=アミノ酸の数):行:違う種類のし
- ペア行列(r*r, r=アミノ酸の数):3次元空間でアミノ酸ペアの関係についての情報
- Structureモジュール 8個
- 原子の三次元空間にある場所を推定する
- Evoformerニューラルネットワークブロック 48個
基礎的な操作一覧
- Linear 重さWの行列+バイアスBで線形変換
- LinearNoBias 重さWの行列だけで線形変換
- LayerNorm Layer normalization
- sigmoid オペレーター
- softmax オペレーター
- stopgrad オペレーター
- ⨀ アダマール積
- ⊗ 直積
- ⊕ outer sum
- aTb 内積
- i, j, k アミノ酸におけるインデックス
- s, t MSAシーケンスにおけるインデックス
- ○ 三次元空間まで変換するオペレーター
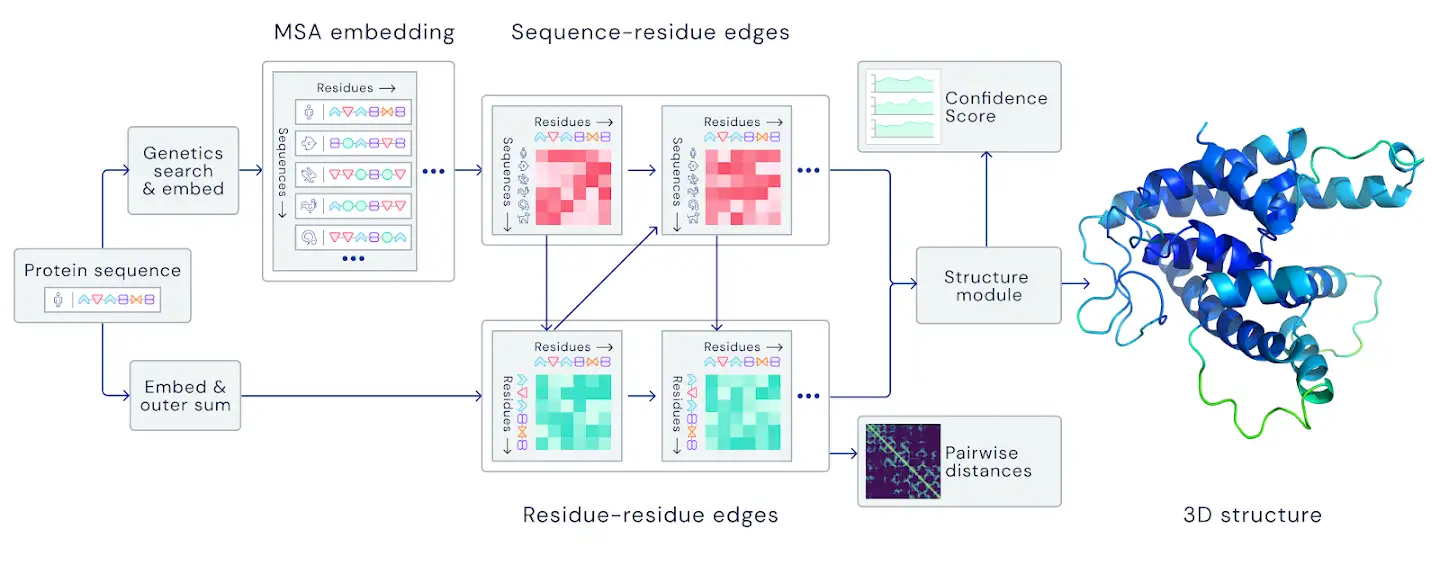
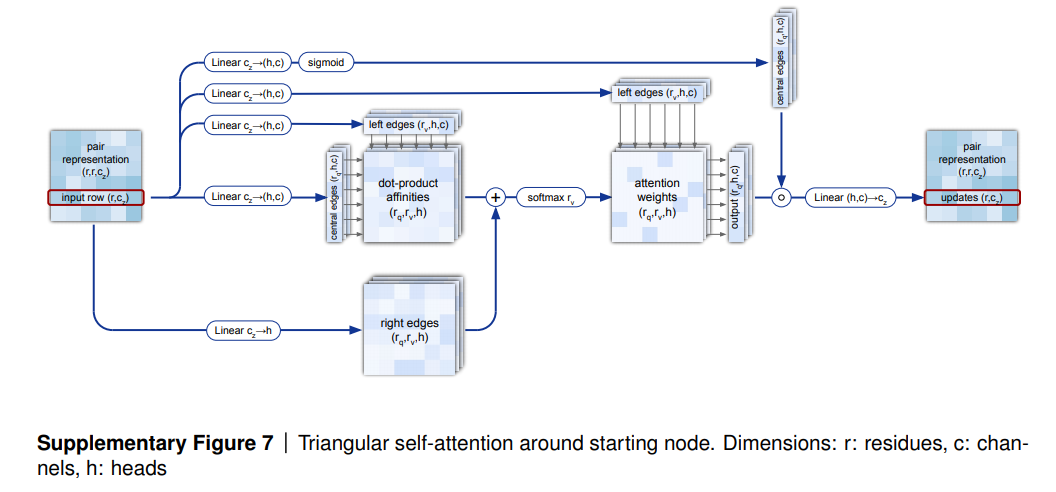
Evoformer
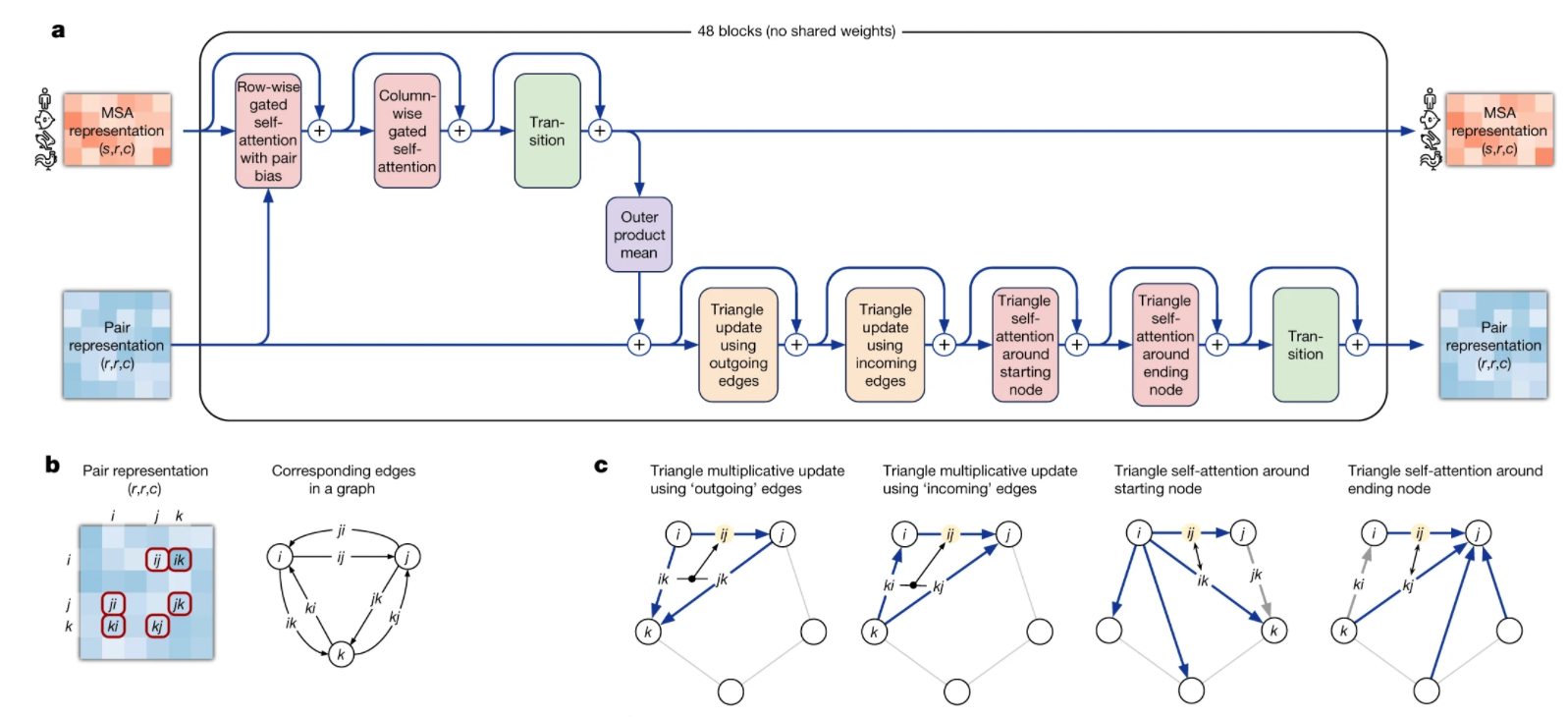
Evoformerの最も重要なアイデアは、ペア行列の中の情報(アミノ酸が三次元空間に関わて情報)をグラフで表すことである。(b)
詳細は以下になる:
- MSA行列の処理において、まずはペア行列を考えながら行ごとにAttentionアップデートを行う, MSAの行における処理によりペア行列・MSA行列の間の相互作用を促進する。
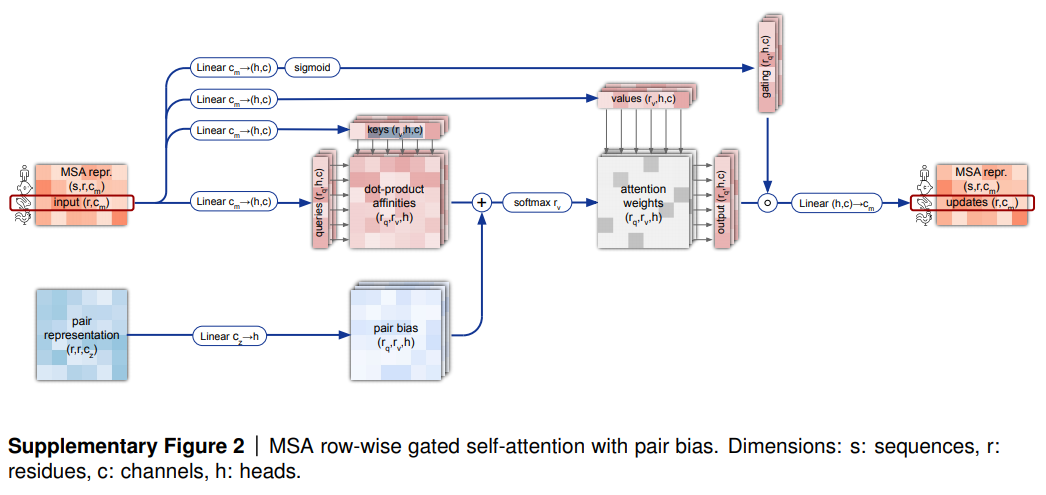
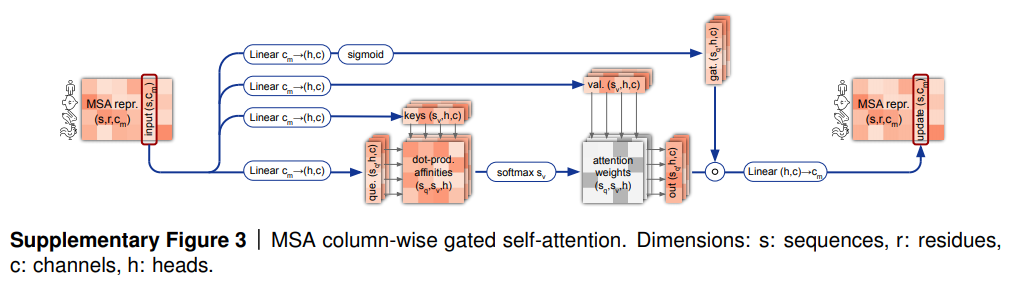
- MSAの列における処理により、同じアミノ酸の進化的な関係の情報を表す。
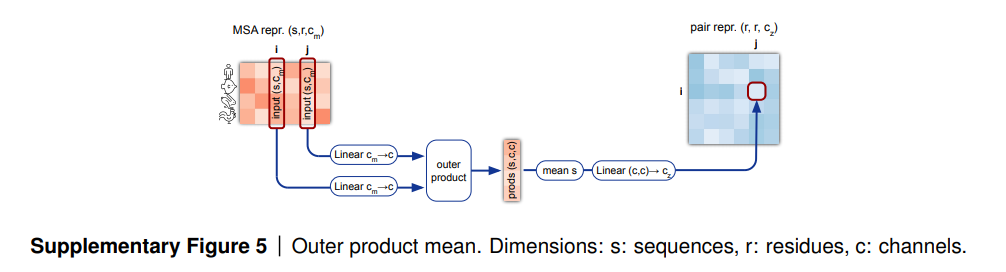
- MSA行列をペア行列に入力すると、列の外積の平均値を取る。この値はペア行列の(i, j)値になる
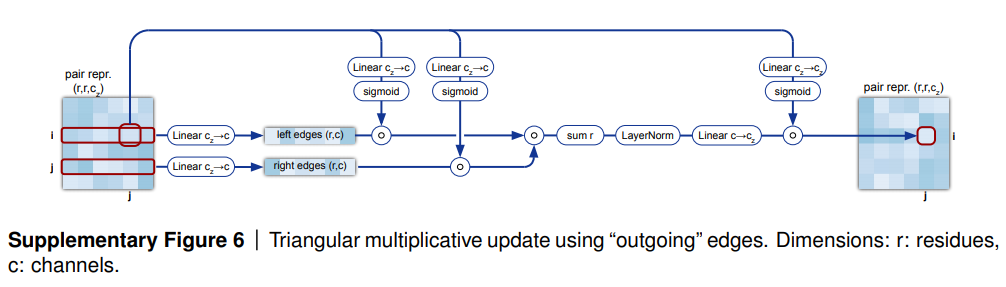
- ペア行列をアップデートすると、いつも縁の三角形ごとにアップデートを行う。つまり、ある縁(i, j)において、i行・j行をアップデートする。また、i列・j列をアップデートするオペレーションもある。上記図3(c)。
- また、 ペア行列をアップデートすると、ある行i(一つのアミノ酸またはグラフの一つのノード)の注意力メカニズムを作る。逆に、列iでも計算できる
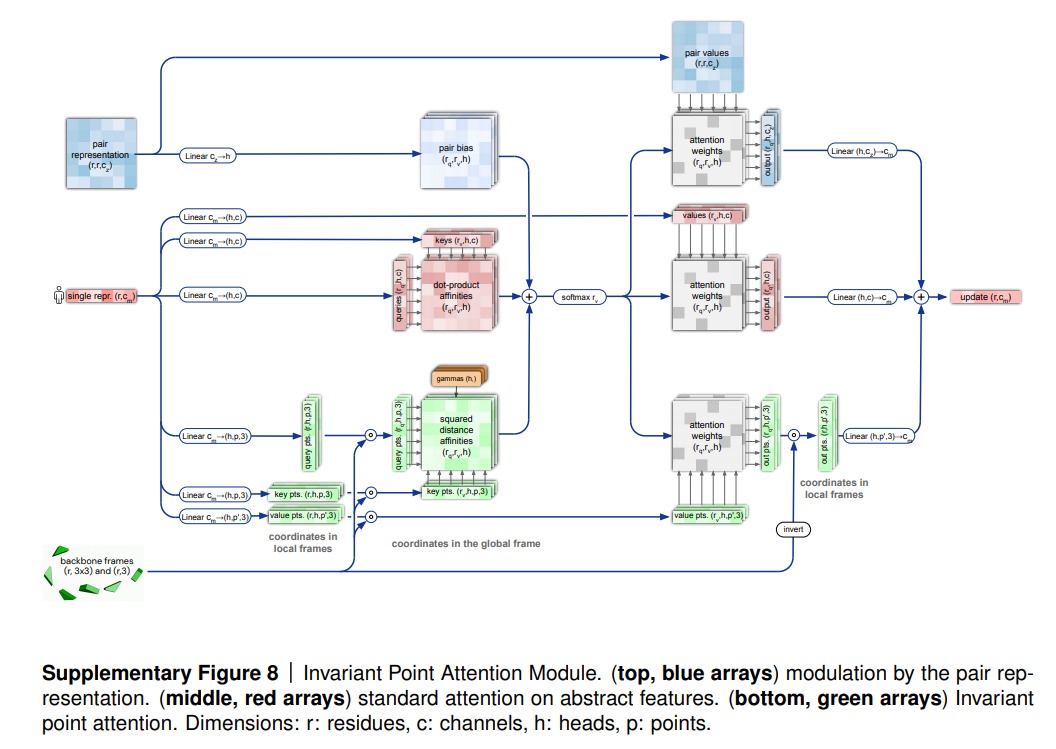
Structure
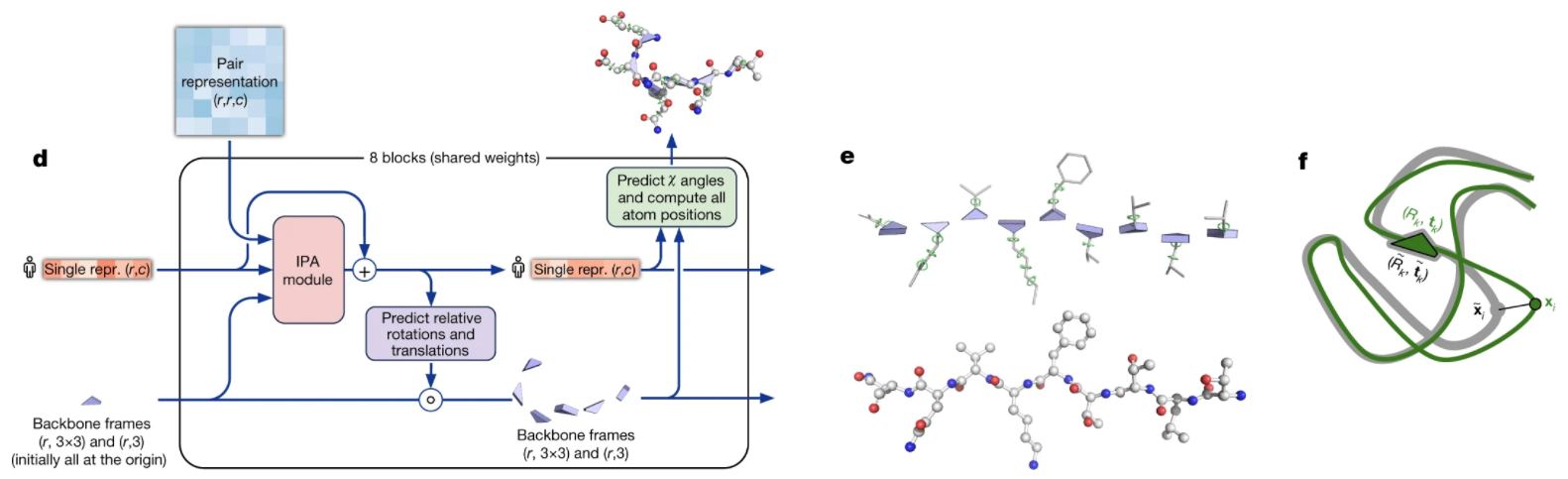
StructureモジュールはEvoformerの抽象的な結果から具体的な三次元モデルを計算する役割を満たす。以下の特徴が持っている:
- 構造:レーヤー数=8とシェアされている重さ。
- 入力:Evoformerの最終的な値を行ごと(一つのアミノ酸)に使い、これをSingle representation {Si} と呼ぶ
- 三次元モデリング:下記の図通り

- IPAとは、invariant point attentionを意味している
- まずはSidechainよりbackboneの場所が優先される
- 最初訓練した時、Sidechainが可能な結合の角度を違反しても一応大丈夫。なぜなら、化学な考慮を無視すると、限局的ン最適化がしやすい。